HIV是引发艾滋病的元凶,曾经人们“谈艾色变”,但是随着对HIV认识的加深以及抗HIV感染药物的研发,艾滋病已从一个令人恐惧的疾病变为可以控制的慢性病。不过,由于不能彻底地清除人免疫缺陷病毒病毒,长期抗病毒治疗仍需面对药物的毒副作用、耐药病毒产生和传播、患者依从性差以及每日口服药物不便利性等棘手的问题。近期出版的《药学进展》“聚焦抗艾滋病药物研发”专题,刊载了四篇颇具特色的综述——《艾滋病抗病毒治疗及药物研究进展》、《抗HIV 天然产物研究进展》、《HIV 进入抑制剂研究进展》、《HIV-1 基因治疗研究进展》,从不同角度总结现今艾滋病治疗与新药研发的进展。我们很荣幸地邀请到国家“千人计划”专家、前沿生物药业(南京)股份有限公司董事长谢东博士为专题点评。
前沿生物由“海归”资深科学家创立于2002年,是一家立足中国、面向全球市场的创新型生物制药企业,专注于研发、生产和销售新药产品,以满足重大临床需求、治病救人、提高人类生活品质。现阶段重点专注于艾滋病抗病毒治疗和疼痛等重要治疗领域创新药物的研发和推广。历经十几年的磨砺和积累,前沿生物已拥有具全球知识产权的长效多肽药物研发平台和多个在研产品,包括具备国际市场竞争力的国家一类抗艾滋病新药艾博韦泰。现分享姚成博士的《艾滋病抗病毒治疗及药物研究进展》。
正文
艾滋病抗病毒治疗及药物研究进展
(前沿生物药业(南京)股份有限公司,江苏 南京210012)
[摘要] 获得性免疫缺陷综合征,又称“艾滋病”,是由人免疫缺陷病毒感染引起的全球流行的传染性疾病。抗逆转录病毒治疗已经取得了显著的临床疗效,但由于不能彻底地清除人免疫缺陷病毒,长期抗病毒治疗需要面对药物的毒副作用、耐药病毒产生和传播、患者依从性差以及每日口服药物不便利性等问题。在目前无**和治愈技术情况下,随着治疗人群迅速扩大、患者寿命及治疗时间显著延长,临床上对艾滋病新药、新技术有持续需求。复方单片制剂、长效注射药物、广谱中和抗体介导的免疫治疗、全新作用机制药物以及预防性**,将是艾滋病防控突破目前瓶颈的关键。综述了艾滋病抗病毒治疗及相关新药的研究进展。
[关键词]人免疫缺陷病毒;获得性免疫缺陷综合征;抗逆转录病毒治疗;新药研发
获得性免疫缺陷综合征(AIDS),又称“艾滋病”,是由人免疫缺陷病毒(HIV)感染引起的全球流行的传染性疾病。自1983 年人类首次发现HIV,迄今已有近4 000 万人因感染HIV 以及艾滋病引发的相关疾病而死亡。联合国艾滋病规划署(UNAIDS)最新信息显示,全球HIV 感染者现有3 670 万,2016 年全球新发感染人数为180 万,约100 万人死于AIDS 相关疾病。
20 世纪90 年代中期,美国FDA 批准非核苷类逆转录酶抑制剂和蛋白酶抑制剂进入临床使用,开启了联合抗逆转录病毒治疗(antiretroviral therapy,ART)的时代。截至2016 年,全球共有1 950 万HIV 感染者接受ART,包括54% 的成人感染者和43% 的儿童感染者。2000—2016 年期间,HIV 新发感染人数下降了39%,与艾滋病相关的死亡人数下降了1/3,约1310 万人因接受ART 而获救。然而,由于ART 无法彻底清除HIV 病毒,长期每日口服多种药物,药物不良反应、病毒耐药以及患者依从性不能保证等问题,艾滋病的治疗依然存在重大的临床需求。
FDA 在1987 年批准了首个核苷类逆转录酶抑制剂(NRTIs)齐多夫定(AZT)。迄今为止,已有29 个抗病毒 药物和多个复方制剂上市。根据药物作用靶点的不同,这些抗病毒 药物主要分为6 大类:NRTIs、非核苷类逆转录酶抑制剂(NNRTIs)、整合酶抑制剂(INSTIs)、蛋白酶抑制剂(PIs)、融合抑制剂(FIs)和CCR5 受体拮抗剂,后两者均属于进入抑制剂(EIs)。本文对抗逆转录病毒 药物的研发历程予以回顾,并对抗HIV 新药研发策略及全球新进展进行综述。
1 抗病毒治疗及药物研发
1.1 “发现即治疗”的全球共识
抗逆转录病毒治疗以维持病毒学抑制、预防病毒传播、减少艾滋病相关疾病、改善生活质量以及延长患者寿命的目标为核心。临床研究表明,HIV 感染早期即开展ART,可以尽早抑制病毒的复制、恢复CD4 细胞正常水平、保护机体免疫功能、降低艾滋病相关疾病的发生率、延缓疾病的进程。HIV 阳性患者接受ART 可以减少病毒传播,有效率达96%,这一概念被称为“治疗即预防”。使用恩曲他滨(FTC)/ 富马酸替诺福韦酯(TDF)对HIV 阴性人群进行暴露前预防,降低HIV 感染的有效率达到92% 。
基于大量的临床研究结果,世界卫生组织(WHO)在2015 年的指南中提出了全员治疗概念,即对全部HIV 阳性感染者进行抗病毒治疗。2016 年美国卫生与人类服务部(DHHS)提出,不论CD4 细胞计数为多少,所有HIV 感染者及艾滋病患者均应尽快开始ART 。2015 年我国修订的《艾滋病诊疗指南第三版》的初始治疗方案中,推荐对CD4 细胞计数在每微升350~500个的感染者“建议治疗”,对CD4 细胞计数高于每微升500 个的感染者“考虑治疗”。2016 年6 月,国家卫计委发布的《关于调整艾滋病免费抗病毒治疗标准的通知》再次明确了“发现即治疗”的原则,建议“对所有艾滋病病毒感染者和患者均实施抗病毒治疗”。
由于不同国家和地区的经济、社会和医疗事业发展不平衡,并不是所有国家和地区都能实现全员治疗。不同国家和地区推荐的一线优选治疗方案也有所差别,DHHS 和欧洲艾滋病临床学会(EACS)指南类似,均为INSTIs 或PIs+2 NRTIs,EACS 还增加了一种利匹韦林(rilpivirine,RPV)/FTC/TDF 方案。WHO 考虑到更广大的发展中国家药物的可及性,推荐的优选方案仍然为NNRTIs+2 NRTIs 。2015 年我国最新的指南推荐的初始治疗方案为NNRTI 或利托那韦(RTV,亦简称r)增强的蛋白酶抑制剂(PI/r)或拉替拉韦(RAL)+2 NRTIs 。
1.2 固定剂量复方制剂
固定剂量复方制剂(fixed-dose combination,FDC)是将多种抗病毒 药物制成单一片剂,可防止AIDS 患者因每日服药数量及次数过多而漏服或者误服,提高其服药依从性。2007 — 2017 年间FDA 共批准了15 个复方制剂,包括:1)基于NRTI 的复方制剂;2)基于NNRTI 的复方制剂;3)基于PI 的复方制剂;4)基于INSTI 的复方制剂。其中,包含完整的治疗药物组合、可以作为单一片剂(single tablet regimen,STR) 用于治疗的FDC 有7 个, 分别是Atripla、Complera(RPV/FTC/TDF)、Stribild、Triumeq、Genvoya、Odefsey(RPV/FTC/TAF)和2017 年最新批准用于维持治疗的两药组合片剂JULUCA(DTG/RPV)。Stribild、Genvoya、Triumeq 和Complera 是DHHS 和EACS 推荐的一线优选方案,Atripla 是WHO 推荐的一线优选方案。
1.3 简化的两药治疗方案
目前为止,所有优先推荐的ART 一线方案均是3个药物的组合,由2 个NRTIs 作为骨架,加上1 个其他类的第3 个药物,如NNRTI、PI、INSTI 或EI。在临床实际应用中,不同国家和地区、不同人群依然面临着治疗费用、药物可及性的挑战。安全性和用药依从性在长期的药物联合治疗中也备受关注。临床和制药界一直在探索更为简单的治疗方案,在保证治疗效果的基础上,采用适当的两药组合方案来减少药物毒副作用,提高用药的方便性和依从性。早期的两药组合方案在有效性和安全性上没有获得足够的数据支持。近年来,随着市场上疗效和安全性更好的抗逆转录病毒新药的出现,两药治疗方案的探索和开发已经取得了可喜的进展。
研究者探索了两药简化方案用于初治人群的可行性。PROGRESS 试验评价了洛匹那韦/ 利托那韦(LPV/r)+ RAL(n=101) 和LPV/r+TDF/FTC(n=105) 在一线治疗中的有效性和安全性,分别有83.2% 和84.8% 患者在治疗48 周时检测不到病毒,两组的不良反应发生率相似,LPV/r+RAL 两药组合不劣于三联疗法。BADAR 试验显示,达芦那韦/利托那韦(DRV/r)+RAL(n=42)作为一线治疗的疗效显著低于三联药物组合DRV/r+TDF/FTC(n=43),分别有62.5% 和83.7% 的患者在48 周时检测不到病毒。然后,另一个大样本试验NEAT 显示了不同的结果,DRV/r+RAL(n=401)和DRV/r+TDF/FTC(n=404)治疗2 年后的失败率分别为19% 和15%,两组的不良反应发生率类似,DRV/r+RAL 两药组合不劣于三联疗法。分层分析显示,在基线高病毒载量和低CD4 细胞人群,DRV/r+RAL 两药组合的有效率更低,同时更容易产生耐药。
除了用于一线初治人群,研究者也在不断探索两药简化方案对已获得病毒学抑制人群维持治疗的可行性。意大利的ATLAS-M 试验入组266 例已获病毒学抑制的HIV-1 感染者,各接受阿扎那韦/利托那韦(ATV/r)+3TC 或ATV/r +2 NRTIs 治疗48 周,分别有89.5% 和79.7% 受试者检测不到病毒。在KITE 研究中,60 名获得病毒学抑制的HIV-1 感染者随机接受LPV/r+RAL 治疗或继续原先的三药方案。治疗48周后两组分别有92% 和88% 的患者维持病毒学抑制。两组均未见发生严重不良事件。对21 个临床随机试验的4 821 个受试者进行的meta 分析显示,尽管总体上会增加耐药发生的风险,一些两药组合用于某些特定人群是安全、有效并且获益的。2015 年WHO 指南已将LPV/r+RAL 二药组合纳入二线治疗的备选方案。FDA 最新批准了DTG 和RPV 的单一片剂Juluca,用于已获得病毒学抑制的HIV-1 感染者的维持治疗。其Ⅲ期临床SWORD-1 和SWORD-2 显示,1 024 例已获得病毒学抑制的HIV-1 感染者按1 : 1 随机分配至DTG/RPV 组或维持当前三联药物治疗,48 周时两组中均有95% 的患者血浆检测不到病毒(HIV-1 RNA < 50 拷贝/mL),DTG/RPV 两药组合不劣于传统的三联疗法。
简化的两药治疗方案可以节省治疗费用,提高用药依从性,提高患者的生活质量,减少不良反应和药物负担,对某些特定人群不失为一种安全有效的选择。
1.4 长效注射药物
随着ART 在临床上的应用和推广,艾滋病已经转变为一个需要终身治疗的慢性疾病。目前已批准的药物均需每日服药,且部分药物需每日服用2 或3 次,给药频率较高,多种药物同时服用将导致药物-药物和药物-食物的相互作用,长期治疗也不可避免地带来毒副作用和产生耐药病毒。除此之外,服药依从性也是严重问题。在接受了多年每日ART 后,许多患者出现治疗疲劳,仅有不到2/3 的患者能够保持获得病毒学抑制所需要的90% 的服药依从性。类似地,尽管WHO 推荐ART 用于暴露前预防,然而,临床试验显示暴露前预防的用药依从性并不太好,存在较大的变化幅度(28% ~ 98%)。当涉及**疾病、药物滥用、经济状况受限、歧视、社会支持差以及治疗方案复杂、药片过多这些合并因素时,长期每天服药的依从性更加成为HIV 感染者和艾滋病患者的巨大挑战。当服药依从性不足时,药物暴露水平将不能够有效抑制HIV 病毒的复制,从而增加了耐药病毒产生的可能性。
长效药物或缓释制剂可为艾滋病的治疗和预防提供新选择,作为一种新策略解决患者依从性差和治疗疲劳的问题。长效药物的定义及最适给药频率尚未取得共识,但可以肯定的是,这些药物都有较长的半衰期,实现方式可以是缓释制剂、药物本身具有较长的半衰期、或是代谢产物具有生物活性。
当前,尚没有长效抗HIV 药物获批准上市,但一系列长效注射药物已进入了后期临床研发阶段,下文将介绍非核苷类逆转录酶抑制剂RPV-LA、整合酶抑制剂Cabotegravir(CAB)、融合抑制剂艾博韦泰(albuvirtide,ABT)、广谱中和抗体(broadlyneutralizingantibodies,bNAbs)3BNC117 等长效药物。这些长效注射药物的上市,尤其是由多个长效药物组成的联合给药方案将为艾滋病的治疗带来又一次的革命性变革,取代每日口服药物的治疗。
1.5 新作用机制药物
HIV 属于逆转录病毒,复制快,每天可产生数百亿新的病毒颗粒。HIV 的变异频率非常高,这是因为逆转录酶缺乏校正修复功能。HIV 耐药可以在接受抗病毒 药物治疗的过程中产生(获得性耐药),也可以是耐药病毒传播到未感染的个体(传播性耐药)。随着暴露前预防和“发现即治疗”的实施,以及ART 在中、低收入国家的普遍推广,因抗病毒 药物广泛、长期叠加应用而导致的获得性耐药和传播性耐药,尤其是NNRTIs 耐药的发生率显著增加。研究证实,超过150 个已知的突变与HIV 耐药有关,抗逆转录病毒治疗6 年的累积耐药性预计可达到27% 。目前在临床上使用的所有药物中,均出现了病毒的耐药性进化现象,甚至包括最新的整合酶抑制剂DTG,其治疗失败的受试者也不可避免地产生了耐药病毒。交叉耐药和多重耐药发生率日益增加,进一步又限制了同类药物中其他品种的使用。
长期的抗病毒治疗对药物的安全性和耐药屏障提出了更高的要求,已发生耐药及缺乏耐受性的患者也需要有更多的药物选择。发现和鉴定新的HIV 药物靶点,探索新的作用机制,研发更多安全、有效、耐受性良好的新药物,为患者提供更多的给药方案,特别是为多重耐药患者提供新的药物选择,将是未来抗HIV药物研发的一个重要方向。近10 年来,整合酶抑制剂和复方固定剂量药物的研发取得极大的成功,尽管尚没有全新作用机制的抗HIV 药物问世,制药企业一直在不断地积极探索,一系列创新药物已经进入临床后期开发阶段,包括下文将介绍的黏附抑制剂(AIs)fostemsavir、成熟抑制剂(MIs)BMS-955176、广谱中和抗体3BNC117 和新一代融合抑制剂ABT 等。预计在不远的将来,多个创新药物将获得批准,丰富抗病毒 药物管线。
2 抗逆转录病毒 药物研发进展及在研新药
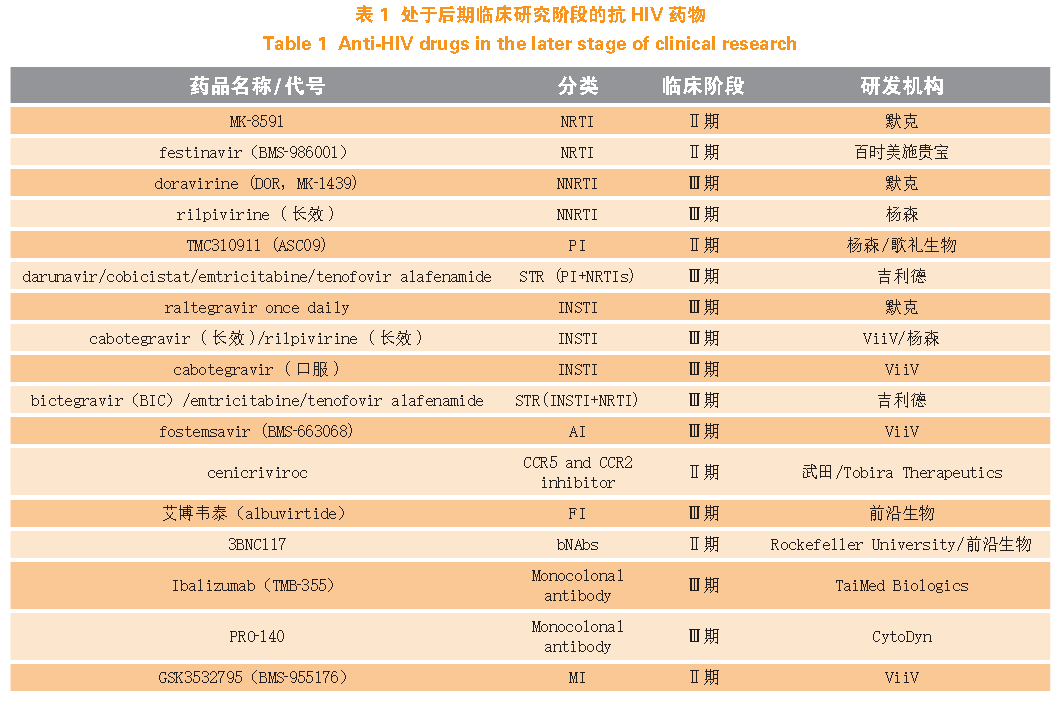
尽管目前可用的抗逆转录病毒 药物数量不断增加,但仍然存在着使用不方便、耐受性差、毒副作用大、容易耐药以及无法清除病毒库等缺点,开发具有新的作用机制的药物具有重大的需求和意义。已有的各大类药物均有正处于研发阶段的新药,其中不乏具有全新作用机制的药物,部分已进入后期临床研究阶段。针对现有药物的劣势,研发的新方向主要是单片复方制剂、长效注射剂、广谱中和抗体及免疫治疗等。长效注射剂和单片复方制剂可减少给药频率和用量,提高依从性和耐受性;广谱中和抗体毒副作用小,可以延迟病毒反弹,激活免疫应答,发挥体液免疫功能。表1 总结了正在后期临床研发阶段的各类抗HIV 新药。
2.1 核苷类逆转录酶抑制剂
NRTIs 是最先被批准的一类抗HIV 药物,通过与逆转录酶竞争性结合,抑制HIV 双链DNA 合成,从而阻止病毒复制。FDA 迄今批准了9 个NRTIs。早期上市的AZT、去羟肌苷(ddI)和司他夫定(d4T)等,由于较严重的线粒体**反应,现已使用较少或不再使用。3TC、FTC、ABC 和TDF 目前仍在临床广泛应用。长期用药的不良反应依然是NRTIs 的主要问题,例如ABC 可引起药物超敏反应,TDF 可造成肾功能障碍和骨代谢异常。TAF 是一种新型的TDF 前体药物,其起效血浆浓度低于TDF,临床上可以给予较低剂量,TAF(10、25 mg)/FTC(200 mg)与TDF(300 mg)/FTC(200 mg)显示了相似的抗病毒活性,从而显著改善TDF 所致的肾小球滤过率降低和骨矿物质密度降低的不良反应。2015—2016 年,多个含有TAF 的复方制剂已获得FDA 批准上市,包括Genvya(EVG/COBI/FTC/TAF)、Odefsey(RPV/FTC/TAF)、Descovy(FTC/TAF)。TAF 长期、大样本量使用的安全性还有待观察,制药领域仍然在积极开发新型NRTIs 药物。
2.1.1 MK-8591
MK-8591 是一种NRTI。MK-8591 与其他NRTIs 不同之处在于它保留了3'-羟基基团,作为易位抑制剂引起无效的链伸长。MK-8591 对HIV-1 和HIV-2 均有活性,包括含NRTI 突变K65R 和Q151M 的病毒。体外试验显示出强大的抗病毒活性,半数有效浓度(EC50)为0.2 nmol · L-1。在猴子模型中,MK-8591磷酸化代谢物的细胞内半衰期约为50 h,显示了每周1次的给药潜力。Ⅰ期临床试验结果显示,单次给药可抑制病毒复制7 ~ 10 d,0.5 ~ 30 mg MK-8591耐受性良好,仅有少量轻中度不良反应发生。给药后7 d,MK-8591 0.5mg 和10 mg 组病毒载量平均下降1.18 log10 拷贝/mL(0.95 ~ 1.46)和1.67 log10 拷贝/mL(1.47 ~ 1.87),30mg 组给药10 d 病毒载量平均下降1.57 log10 拷贝/mL(1.34 ~ 1.85)。评价MK-8591 与DOR 和3TC 联用在初治HIV 感染者中的耐受性、抗病毒活性和药动学的Ⅱ期临床试验(NCT03272347)正在进行中。
2.1.2 Festinavir
Festinavir 是一种新型NRTI, 它是与d4T 相似的胸苷结构类似物。体外研究显示,festinavir 能抑制宿主DNA 合成,而不影响线粒体功能,具有较高的安全性。多数携带M41L、D67N 和K103N 耐药突变(无论是否有M184V 突变)的病毒对festinavir 的敏感性降低,但携带Q151M 和K56R 突变的多重耐药病毒对festinavir 则是轻度超敏。Ⅰ/Ⅱ期临床试验结果显示,已接受过ART 治疗的HIV-1 感染者接受festinavir 100 ~ 600 mg 单药治疗10 d,病毒载量降低的中位数显著高于安慰剂组。Ⅱ期临床剂量探索研究正在进行中,将比较festinavir+3TC +EFV 和TDF+3TC+EFV 两组治疗48 周的有效性和安全性。
2.2 非核苷类逆转录酶抑制剂
NNRTIs 直接与逆转录酶活性位点结合,降低酶活性,从而抑制病毒复制。奈韦拉平(NVP)、地拉韦啶(DEL)和EFV 是FDA 批准的第1 代NNRTIs,依曲韦林(ETR)和RPV 是第2 代NNRTIs。NNRTIs 具有很强的抗病毒活性,但其临床应用受到不良反应、药物相互作用和耐药屏障较低的限制。NNRTIs 通过CYP450 代谢,主要不良反应包括中枢神经系统(CNS)症状、肝**、皮疹、抑郁、失眠、躁狂等**障碍等。NNRTIs 的耐药屏障较低,容易产生交叉耐药,是目前耐药率的一类抗HIV 药物。尽管存在以上缺陷,NNRTIs 仍然是WHO 推荐的初始治疗药物,与2 种NRTIs 组成联合抗病毒治疗方案。2011 年FDA 批准的RPV 具有较高的抗病毒活性,半衰期更长,不良反应较轻,耐药屏障相对较高,但限制应用于病毒载量低于100 000 拷贝/mL 的患者。一些预期在疗效、安全性、耐药屏障方面更好的NNRTIs 先导化合物和试验药物也在开发之中。
2.2.1 Doravirine
DOR 是新一代NNRTI。临床前体外试验显示,DOR 具有很强的抗HIV-1 活性和独特的耐药特征,与其他NNRTIs 之间的交叉耐药有限。DOR对常见的K103N、Y181C、G190A、E101K、E138K和K103N/Y181C 耐药突变病毒具有抗病毒活性,而使用DOR 进行体外耐药病毒培养,选择出的耐药突变为V106A、F227L 和L234I,不同于其他NNRTIs引起的突变。与其他NNRTIs 相比,DOR 具有每日服药1 次、不抑制或诱导CYP450 酶、没有显著的食物影响等特征。
Ⅱ期临床试验结果表明,DOR+FTC/TDF 与EFV+FTC/TDF 的临床疗效相似。在第1 部分剂量筛选阶段,患者被随机给予25、50、100 或200 mgDOR 或600 mg EFV 治疗。在第24 周时,所有接受DOR 的患者都将剂量调整为100 mg。治疗48 周,DOR 组77.8%(84/108)患者HIV-1 RNA < 40 拷贝/mL,而EFV 组为78.7%(85/108)。2017 年法国巴黎HIV 科学国际大会(IAS 2017)报告了在初治HIV-1感染者中开展的Ⅲ期临床试验DRIVE-AHEAD 的结果,相较于EFV/FTC/TDF 固定剂量复方,每日1 次的DOR/3TC/TDF 的固定剂量复方片剂达到了主要的非劣性疗效终点。治疗48 周,DOR/3TC/TDF 组有84.3%(307/364)的受试者HIV-1 RNA < 50 拷贝/mL,而EFV/FTC/TDF 对照组这一比例为80.8%(294/364),相差3.5%,95%CI(-2.0,9.0)。DOR 组中枢神经系统不良反应眩晕、睡眠障碍和感觉异常的发生率均显著低于EFV 组,具有统计学意义(P < 0.001,P < 0.001和P=0.033)。
2.2.2 Rilpivirine-LA
RPV 是已上市的第2 代NNRTI,具有相对良好的安全性和高耐药屏障。RPV 每日1 次给药的口服制剂已经使用数年。近年来,研究人员正在开发RPV 长效注射制剂(RVP-LA),其为晶体纳米混悬剂。RPV-LA 肌肉注射给药后6 ~ 8 d 内达到血药浓度峰值,消除半衰期为44 ~ 61 d,18 个月后仍然能在血浆和女性生殖液中检测到药物,但低于有效治疗浓度。RPV 整体暴露量与性别和体重指数(BMI)有关,女性的峰值血浆浓度比男性低大约30%,并且BMI 每增加1 kg · m-2,峰值血浆浓度大约下降2.3%,BMI 高于25 kg · m-2 的女性可能出现低峰值浓度的风险。研究者未观察到性别或BMI 对于服药28 d 后药物血浆浓度的影响。
2.3 蛋白酶抑制剂
PIs 通过抑制蛋白酶的水解活性,阻止病毒前蛋白的裂解和形成成熟的感染性病毒粒子。迄今为止,FDA批准了10 个PIs。第2 代PIs LPV、ATV 和DRV 与药代增效剂RTV 联合使用,均显示较好的临床疗效。PIs具有较强的抗病毒活性和较高的耐药屏障,但存在特异性不良反应,包括血脂异常、胰岛素抵抗、高血糖、脂肪代谢障碍。此外,PIs 主要经P450 酶代谢,是CYP3A4 的抑制剂,药物相互作用较为常见,还可能增加HIV 阳性的血友病患者的出血风险。因此,随着长期用药带来越来越多的交叉耐药、显著的药物相互作用和不良反应,也需要开发新的PIs 以满足临床需求。
2.3.1 TMC310911
TMC310911 是新一代的PI。体外试验显示,TMC310911 对多种HIV-1 临床分离株及耐药株具有抗病毒活性。与DRV 或LPV 相比,TMC310911 可以减少耐药毒株的产生,降低病毒突变的发生率,表明TMC310911 比现有其他PIs 具有更高的耐药屏障。临床Ⅰ~Ⅱa 期试验结果显示,TMC310911 的安全性和耐受性良好。除了胃肠道不良反应外,接受TMC310911 治疗的健康受试者中未见严重并发症。TMC310911 具有线性药动学特征,且与RTV 联用可提高其生物利用度。Ⅱa 期临床试验显示,TMC310911 与RTV 联用在初治HIV-1 感染者中显示出强效的抗病毒活性,治疗2 周后血浆HIV-1 RNA 降低超过1.5 log10 拷贝/mL,且在所有治疗剂量下安全性良好。基于Ⅱa 期试验数据,目前研究者正在开展进一步的临床研究(NCT00838162)。
2.3.2Darunavir/Cobicistat/Emtricitabine/Tenofovir Alafenamide
DRV 800 mg/COBI 150 mg/ FTC200 mg/TAF 10mg 是一个以PI 为核心的复方单一片剂,每日1 次口服给药,目前正在进行Ⅲ期临床试验和生物等效性研究。如果被批准,这将是第1 个包含PI 的STR。在一项多中心、随机、双盲、阳性对照的Ⅱ期临床试验中,研究人员比较了每日1 次DRV 800 mg/COBI 150 mg/FTC200 mg/TAF 300 mg(基于TAF 方案)与DRV 800mg+COBI 150 mg+ FTC 200 mg/TDF 300 mg(基于TDF方案)在153 例初始治疗且肾小球滤过率(eGFR)不低于70 mL · min-1 的HIV-1 成年感染者中的有效性和安全性。试验达到非劣效临床终点,在48 周时,基于TAF 方案的病毒学抑制率是76.7%,而基于TDF 方案的病毒学抑制率是84.0%。在48 周时,TAF 和TDF 组分别各有36 例受试者病毒学治疗失败,但没有产生耐药。在第48 周,TAF 组显示了更好的肾 脏和骨骼安全性。TAF 组和TDF 组血清肌酐(SCr)的平均变化分别为0.6与0.9 mg · L-1(P=0.053),髋骨密度(BMD)的变化百分比为-0.84% 和-3.82%(P < 0.001),脊柱骨密度(BMD)的变化百分比为-1.57% 和-3.62%(P=0.003)。与目前首选初治药物DRV/r 相比,该新配方STR 可以减少药物负担,改善肾 脏和骨骼的安全性。
2.4 整合酶抑制剂
INSTIs 又称整合酶链转移反应抑制剂,通过竞争性结合整合酶的活化位点,抑制病毒cDNA 整合入宿主基因组的链转移环节,从而阻断HIV 病毒复制。FDA 相继批准了3 个INSTIs——RAL、EVG 和DTG,分别于2007、2012 和2013 年上市。INSTIs 抗病毒疗效显著,可以快速降低HIV RNA,且一般都有很好的耐受性,已成为发达国家一线治疗方案的首选药物。其中EVG 与增效剂COBI 联合使用可增强抗病毒疗效。第1 代INSTIs 的RAL 和EVG 耐药屏障较低,1 ~ 2 个突变即降低了病毒对RAL 和EVG 的敏感性,且RAL与EVG 存在较高的交叉耐药性。第2 代INSTIs 的DTG 有较高的耐药屏障,对约90% 的RAL 和EVG 耐药病毒仍然有效,自2013 年上市以来得到广泛应用,已被DHHS 推荐为一线治疗药物。近年来,已出现DTG 病毒学治疗失败的病例报道,发现的耐药相关突变包括R263K、N155H 和S230R 。整合酶是抗HIV 治疗的已被验证靶点之一,多个新的整合酶抑制剂处于临床开发之中。
2.4.1 Raltegravir
RAL 是一种已上市的INSTI,每日2次给药,是未接受过抗病毒治疗的HIV-1 患者首选一线方案中的药物。为了简化基于RAL 的用法,目前正在开发每日1 次的给药方案。QDMRK 试验中,研究者评估了每日1 次的给药策略,未能证明每日1 次800mg RAL 不劣于已批准的每日2 次400 mg RAL。目前,研究者正在评估比较RAL 1 200 mg(给予2 片600 mg片剂)每日1 次与标准治疗每日2 次给药在初治患者中的疗效。初步数据表明,与FTC 200 mg/TDF 300 mg联用,每日1 次RAL 1 200 mg 不劣于每日2 次RAL400 mg 。RAL 每日1 次给药方案可为HIV-1 感染者提供另一种优选的一线治疗选择,减少药物相互作用。
2.4.2 Cabotegravir
CAB 是一种新的INSTI,结构上类似于DTG,在治疗HIV-1 感染方面具有相似的耐药性。CAB 被制成每日服用的口服片剂以及基于其长半衰期的每月或每季度皮下或肌内给药的长效纳米悬浮剂。与EVG 不同,CAB 不需要增效剂,因此没有CYP3A4 药物相互作用,主要通过UGT1A1 代谢。
LATTE-2 是一项在初治HIV-1 感染者中开展的平行、开标记的Ⅱb 期临床研究。受试者先接受20 周的诱导期治疗,每日口服30 mg CAB+3TC/ABC(在诱导期最后4 周口服25 mg RPV)。治疗20 周后,获得病毒学抑制(血清HIV-1 RNA < 50 拷贝/mL)的患者进入维持期治疗,按照2 : 2 : 1 随机分为3 组,分别是间隔4 周肌注400 mg CAB 和600 mg RPV(4 周组)、间隔8 周肌注600 mg CAB 和900 mg RPV(8 周组)或持续保持口服方案30 mg CAB+ABC/3TC(口服组)。研究结果显示,309 例患者中的286 例在诱导期获得病毒学抑制,4 周组、8 周组和口服组分别有94%(108/115)、95%(109/115)和91%(51/56)的患者维持病毒学抑制。治疗96 周时,4 周组、8 周组和口服组分别有87%(100/115)、94%(108/115)和84%(47/56)的患者维持病毒学抑制。仅有8 周组2 例和口服组1 例受试者发生病毒学治疗失败。共230 例受试者接受4 360 次注射,轻度和中度注射位点反应发生率分别为84% 和15%,仅有2 例因注射反应而终止试验,注射点疼痛经常出现。维持期肌肉注射两组共发生严重不良事件22例(10%),口服治疗组发生7 例(13%),但均与药物无关。
上述试验结果表明,CAB 和RPV 这2 个长效制剂联合,间隔4 周或8 周用药,维持治疗已获得病毒学抑制的HIV-1 感染者的疗效明显,而且依从性和耐受性良好。ViiV 和杨森公司合作,已启动2 项Ⅲ期临床研究FLAIR 和ATLAS,将分别评价每月1 次的双药注射方案在初治和已接受ART 治疗的HIV-1 感染者中的有效性和安全性。
2.4.3 Bictegravir
BIC 是一种新的第2 代INSTI,每日使用1 次,且无需药代增效剂COBI。BIC 在健康人群中耐受性良好,可抑制肾小管转运蛋白,降低肌酐水平,不损伤肾功能。与其他整合酶抑制剂相比,BIC 具有更高的耐药屏障和更少的药物相互作用。
2017 年在波士顿的逆转录病毒和机会性感染会议(CROI)上,研究者公布了正在开展的一项Ⅱ期临床试验结果,在用于初治的HIV-1 感染者的一线方案中,BIC 方案与DTG 方案的疗效相似,且具有良好的安全性和耐受性。目前吉利德公司正在进行BIC/FTC/TAF单一片剂的Ⅲ期临床开发,开展了4 个Ⅲ期临床研究,用于初治以及获得病毒学抑制后更换治疗方案的成年感染者。结果显示,在全部4 个试验中,BIC/FTC/TAF方案均达到了非劣效性的主要临床终点,BIC/FTC/TAF具有良好的耐受性,无受试者因肾 脏不良事件停药。吉利德公司于2017 年6 月向美国FDA 和欧洲药品管理局(EMA)提交了BIC/FTC/TAF 治疗HIV-1 成年感染者的新药申请(NDA)和上市许可申请(MAA)。
2.5 融合抑制剂
融合抑制剂(EIs)是一大类新的抗逆转录病毒 药物,作用于病毒生命周期最早期的关键环节,干扰HIV 与宿主细胞的黏附或融合,阻断HIV 进入细胞。HIV 病毒粒子进入靶细胞经历了病毒黏附到细胞表面、与CD4 受体及辅助受体结合、gp41 蛋白介导的病毒-细胞膜融合3 个步骤。根据作用步骤的不同,EIs 又可以分为AIs、辅助受体结合抑制剂(CCR5 或CXCR4拮抗剂)和融合抑制剂(FIs)。EIs 是近年来主要研究方向之一,目前有多个全新作用机制EIs 正处于临床研究阶段。
恩夫韦肽(T20)是FDA 最早批准的融合抑制剂,也是目前唯一批准上市的融合抑制剂,通过抑制HIV病毒包膜与细胞膜的融合而阻断病毒进入宿主细胞。因其独特的作用机制,T20 对其他抗逆转录病毒 药物的耐药病毒有效,临床上对多重耐药患者疗效显著。T20 是唯一的非口服药物,给药方式为皮下注射给药。但由于需每天2 次皮下注射,绝大部分患者出现不同程度的注射位点反应,以及超敏反应和肺炎发生率增加,T20 临床使用受到了一定的限制,常用于多重耐药感染者的治疗。
ABT 是一种新的第2 代融合抑制剂,其作用靶点为gp41 上一个新的保守区域,与T20 的作用区域不同。ABT 是由34 个氨基酸组成的化学合成多肽,其多肽序列衍生自HIV-1 gp41 的N-末端序列,第13 位赖氨酸侧链含有3-马来酰亚胺基丙酸(MPA),修饰后该多肽在血液里能够迅速与白蛋白发生1 : 1 特异结合,形成稳定共轭体,延长其体内半衰期。体外试验显示,ABT 对各种病毒亚型及耐药病毒株均有较强的抗病毒活性,且与现有抗HIV 药物(包括T20 在内)均无交叉耐药性。ABT 与AZT 和沙奎那韦(SQV)具有协同作用,与EFV 和T20 表现为相加作用。ABT 具有较高的耐药屏障。
Ⅰ期临床试验显示,ABT 在HIV-1 感染者中显示出良好的耐受性和安全性,以及明确的剂量相关的抗病毒活性,半衰期11 ~ 12 d,单次给药可以抑制病毒6 ~ 10 d 。Ⅱ期临床试验显示,ABT(160 或320 mg每周1 次静脉给药)和LPV/r 两药组合在初治HIV-1感染者的安全性良好,具有确定的抗病毒疗效,在治疗第7 周,160 和320 mg ABT 剂量组HIV-1 RNA 分别较基线下降了1.9 log10 拷贝/mL(1.3 ~ 2.3)和2.2log10(1.6 ~ 2.7)拷贝/mL 。Ⅲ期临床试验TALENT是一项为期48 周的随机、开标记、非劣效性试验,在一线ART 治疗失败的HIV-1 感染者中开展。受试者按1 : 1 比例分别接受ABT(每周1 次静脉给药)和LPV/r 两药组合,或者WHO推荐的二线三药组合(LPV/r + TDF 或AZT + 3TC)治疗。中期分析显示,治疗48 周,试验组和对照组主要疗效指标——HIV-1RNA < 50 拷贝/mL 的受试者百分率分别为80.4% 和66.0%,组间差值的双侧95%CI 为-3.0% ~ 31.9%,试验组不劣于对照组,其他3 个次要疗效指标均达到方案预设终点。不良事件发生率无组间差异,未发生药物相关的严重不良事件,ABT 每周1 次静脉给药,未观察到注射位点反应。基于Ⅲ期临床的有效性和安全性结果,前沿生物已向CFDA 递交了新药注册申请。
2.6CCR5 受体拮抗剂
马拉维诺(maraviroc,MVC)是目前FDA 唯一批准的CCR5 受体拮抗剂,通过抑制CCR5 辅助受体与病毒包膜蛋白gp120 的结合而阻断病毒进入宿主细胞。在临床使用中已报道的不良反应有肝**、上呼吸道感染、发热和体位性低血压。由于需要的辅助受体不同,HIV-1 病毒又分为CCR5 嗜性和CXCR4 嗜性,且在感染过程中存在病毒嗜性的转化,MVC 对CXCR4 嗜性病毒无效。MVC 在使用前或治疗失败后需要做病毒嗜性检测,这限制了其临床使用,尤其在低、中等收入国家。
Cenicriviroc 是一种在研的CCR5 受体拮抗剂。虽然cenicriviroc 与MVC 一样都需要在治疗前进行病毒嗜性检测,但不同的是,cenicriviroc 可以同时拮抗CCR5 和CCR2 受体。在一项双盲、安慰剂对照的Ⅱ期临床研究中,感染CCR5 嗜性病毒并已接受过ART 治疗的HIV-1 感染者,接受cenicriviroc 单药治疗10 d 后,显示了良好的安全性和有效性。
另一项Ⅱb 期临床试验中, 研究者评价了cenicriviroc 100 mg 或200 mg+FTC/TDF 和EFV+FTC/TDF 在初治HIV-1 感染者中的有效性和安全性。试验结果显示,在143 例受试者中,治疗48 周时,cenicriviroc 100 mg 组、200 mg 组和EFV 组分别有68%、64% 和50% 受试者达到病毒学抑制(P > 0.05,与EFV 组比较)。5 例接受cenicriviroc 治疗的受试者产生耐药,而EFV组没有耐药发生。与EFV 相比,cenicriviroc 组与治疗相关的2 级不良反应(P=0.002)和导致终止治疗的不良反应(P < 0.001)发生率较少。基于以上数据,研究者将选择cenicriviroc 200 mg 进行Ⅲ期临床研究。
2.7 黏附抑制剂
Fostemsavir(BMS-663068)是一种新型进入抑制剂,作用于病毒进入细胞的起始阶段的黏附环节,具有该作用机制的药物也被称为黏附抑制剂(AIs)。迄今尚无AIs 批准上市。Fostemsavir是temsavir 的一种前体药物,temsavir 通过与HIV-1 gp120 直接结合,阻断病毒的初始附着,防止其进入宿主CD4 细胞。由于其独特的作用机制,fostemsavir 可能为耐药患者提供更多的选择。
在一项随机对照的Ⅱb 期临床试验中,251 例已接受过ART 治疗的受试者按照1 : 1 : 1 : 1 : 1 的比例随机分为5 组,fostemsavir 4 个剂量组(400 mg 和800 mg,每日2 次;600 mg 和1 200 mg,每日1 次)和对照组(每日1 次300 mg/100 mg ATV/r),均与每日2 次400 mgRAL+ 每日1 次300 mg TDF 联用。试验结果显示,治疗48 周时,fostemsavir 各组与ATV/r 组疗效相似,fostemsavir 组HIV-1 RNA < 50 拷贝/mL 的受试者百分率为61% ~ 77%,ATV/r 组为71%。在治疗的剂量范围内,fostemsavir 耐受性和安全性良好,未见因药物相关的不良反应而导致的停药。
一项针对多重耐药人群的Ⅲ期临床试验正在进行中。受试者为已接受过ART 治疗的HIV-1 感染者,且至少对3 类药物产生耐药、不耐受和/或有禁忌证。根据受试者能够获得的可耐受且仍然保持病毒学敏感的药物的数量,将受试者分配到随机或非随机的队列,有1 或2 个活性药物可供选择的患者被分配到随机队列,没有任何活性药物可以选择的受试者被分配到非随机队列。随机化队列中的患者在现有ART治疗方案的基础上,每日2 次口服安慰剂或600 mgfostemsavir,从第9 d 开始,全部受试者每日2 次口服600 mg fostemsavir 加上优化背景方案(OBR)。非随机队列患者将在第1 d 开始接受每日2 次口服600 mgfostemsavir+OBR。全部患者接受48 周疗程治疗,预计在2018 年获得试验结果。
2.8 广谱中和抗体
HIV 的高频变异和强大的免疫逃逸能力,导致机体不能产生有效的免疫应答,大量的**研究因此失败。bNAbs 是指一类能够中和多种病毒亚型的抗体,约20% 的HIV-1 长期感染者在2 ~ 3 年后能产生抗HIV-1 的bNAbs。但是,经自然途径或者传统的人工免疫均无法有效地诱导HIV-1 感染者产生bNAbs,研究者开始尝试用分离到的bNAbs 进行被动免疫治疗。近年来,随着B 细胞培养和单克隆抗体技术的突破和改进,从HIV 感染者记忆型B 细胞中分离并构建的新一代bNAbs 的成功概率和数量大大提高,已获得100多种bNAbs。体外试验显示,bNAbs 可以阻断病毒在细胞间的传播,动物试验也证实bNAbs 给药后的被动免疫可以抑制病毒感染,延迟或阻止血浆病毒反弹。在多个临床试验中bNAbs 也显示出明确的保护效果。bNAbs不但能中和游离的病毒颗粒而发挥抗病毒作用,还可以有效地刺激机体免疫系统,促进感染者的体液免疫应答,加速清除被病毒感染的细胞。除此之外,研究者还在积极尝试,利用广谱中和抗体进一步探索无药缓解(drug-free remission)或功能性治愈(functionalcure)。发现并开发有效的bNAbs 成为近年来抗HIV研究的重要方向和热点。
就抗病毒作用机制来说,广谱中和抗体也是HIV进入抑制剂,作用于病毒进入细胞起始阶段。根据识别和作用位点的不同,抗HIV-1 的bNAbs 主要分为以下几类:1)针对病毒gp120 的CD4 结合位点(CD4bs),例如:VRC01、3BNC117 和10-1074 等;2)针对病毒V1V2 位点,例如:PG9 和PG16 等;3)针对病毒保守的V3 位点,例如:PGT121 和PGT128 等;4)针对病毒gp41 的MPER 区, 例如:4E10 和10E8 等;5)针对病毒gp120 上结合趋化因子受体的位点(CD4iAb),例如:17b 和E51 等。3BNC117 是目前临床开发进度最快的bNAb 之一。
3BNC117 是一种lgGlκ 同型的重组全人单克隆抗体(mAb), 可与HIV-1 包膜蛋白gp120 的CD4 结合位点发生特异性结合。体外试验显示,3BNC117对HIV-1 具有广谱高效的中和活性,能够中和237 株HIV-1 病毒中的195 株,包括6 个不同的亚型,平均IC50 为0.08 mg · L-1。在Ⅰ期临床试验中,12 例健康受试者和17 例HIV-1 感染者分别接受1、3、10、30 mg ·kg-1 3BNC117 单次静脉滴注。3BNC117 各剂量组均显示了良好的安全性和耐受性。3BNC117 在HIV-1 感染者体内的半衰期为9.9 d,在健康受试者体内的半衰期为17.5 d。30 mg · kg-1 3BNC117 单次给药,病毒载量平均下降1.48 log10 拷贝/mL(0.8 ~ 2.5),并维持较低水平28 d 。3BNC117 介导的免疫治疗可以增强宿主的体液免疫反应,几乎所有接受3BNC117 静脉滴注的受试者对异源病毒的中和活性都得到了显著改善。研究显示,3BNC117 不仅清除游离的病毒,阻止新的感染,还可以加速清除被感染的细胞。3BNC117 靶向感染病毒的CD4 细胞,并通过Fcγ 受体参与的机制来发挥免疫清除作用。
在一项Ⅱ期临床试验中,已获得病毒学抑制的HIV-1 感染者接受每3 周1 次、共2 次,或者每2 周1 次、共4 次的30 mg·kg-1 3BNC117 静脉滴注,同时停止口服药物治疗。结果显示,每3 周1 次共2 次滴注后病毒反弹时间延迟5~9 周,平均6.7 周,每2 周1 次共4次滴注后病毒反弹最长延迟19 周,平均9.9 周。而历史数据显示,停用口服药物后,病毒平均反弹时间为2.6周(P < 0.000 01)。上述研究显示,3BNC117 可以抑制病毒复制,延迟病毒反弹,诱导人体免疫应答。
2.9 其他治疗性单克隆抗体
2.9.1 Ibalizumab
Ibalizumab 是一种人源化单克隆抗体,其作用机制为通过结合CD4 受体的细胞外结构域Ⅱ,引起后者构象变化,阻止病毒融合及进入CD4 细胞。体外试验显示,ibalizumab 的IC50 为0.03 mg · L-1,能够中和92% 的HIV-1 病毒。早期临床试验结果表明,在给药后14 d, 接受单剂量静脉注射10 mg · kg-1ibalizumab 的受试者,HIV-1 RNA 水平降低1.33 log10拷贝/mL。
目前,研究者开展了Ⅲ期临床试验,在已接受抗病毒治疗的多重耐药HIV-1 成年感染者中评价ibalizumab的安全性和有效性。入组受试者在第-6 d 继续接受当前失败的ART 方案治疗,并在第7 d 随机接受安慰剂或单次静脉注射ibalizumab 2 000 mg。从第14 d 开始,全部受试者给予优化背景方案(OBR)。从第21 d 开始,患者每2 周接受1 次静脉注射(ibalizumab 800 mg),直至第23 周。主要疗效终点是ibalizumab 治疗开始后7 d(研究的第14 d)HIV-1 RNA 下降不低于0.5 log10 拷贝/mL 的受试者比例。研究结果显示:治疗第14 d 试验组和对照组病毒载量下降不低于0.5 log10 拷贝/mL 的受试者比例分别为83% 和3%(P < 0.000 1),试验组病毒载量平均降低1.1 log10 拷贝/mL(P < 0.000 1)。给药24 周后,与基线相比,病毒载量平均下降1.6log10 拷贝/mL;分别有55% 和48% 的患者病毒载量下降大于1 log10 和2 log10 拷贝/mL;43% 的患者病毒载量达到检测限以下,50% 的患者病毒载量低于200 拷贝/mL。药物安全性良好,共9 例患者报告了17 例严重不良反应(SAE),其中仅1 例与药物相关的停药,9 例与药物无关的停药,4 例死亡,3 例撤回知情同意,2 例失访;无受试者产生抗药抗体。目前TaiMedBiologics(中裕新药)已向FDA 提交了ibalizumab 的生物制品许可申请(BLA),FDA 已受理并授予其优先审评资格。
2.9.2 Pro140
Pro140 是一种人源化单克隆抗体,靶向宿主细胞第二受体CCR5,阻止病毒进入细胞。体外实验显示,Pro140 与小分子CCR5 拮抗剂有协同作用和有限的交叉耐药性。在短期单一疗法研究中,研究人员评价了Pro140 静脉和皮下注射对CCR5 病毒嗜性的HIV-1 感染者的活性。与安慰剂相比,Pro140 的2 种剂型均具有良好的耐受性,并且表现出强效、延长的和剂量依赖性的抗病毒活性。
在Ⅰ期研究中,受试者接受0.5、2 和5 mg · kg-1Pro140 单次静脉注射,抗病毒活性呈剂量依赖性增加,在5 mg · kg-1 剂量下HIV-1 RNA 平均降低1.83 log10 拷贝/mL。一项随机、双盲、安慰剂对照、平行的Ⅱ期临床研究共入组31 例CCR5 病毒嗜性的HIV-1 感染者,以1 : 1 : 1 随机分配至5、10 mg · kg-1 Pro140 组和安慰剂组,单次给药静脉注射,随访至治疗后58 d,观察抗病毒活性、耐受性和药动学。研究结果显示,与基线相比,5 mg · kg-1 和10 mg · kg-1 组HIV-1 RNA 平均降低1.8 log10 拷贝/mL(与安慰剂组相比,P < 0.000 1)。Pro140 2 个剂量组的病毒载量在给药12 d 后达到点,且29 d 仍保持低水平,与安慰剂组差异显著(P< 0.01)。Pro140 体外受体嗜性和病毒敏感性分析显示,接受Pro140 治疗的全部受试者均维持CCR5 受体嗜性,除了10 mg · kg-1 组中的1 例受试者在第15 d 观察到双重/混合病毒。所有病例的抑制率在治疗前后无变化(治疗前≥98%,治疗后≥ 99%)。Pro140 不抑制CXCR4 介导的双重/ 混合病毒进入U87-CD4-CXCR4细胞。Pro140 安全性良好,未报告SAE 或剂量限制性**,不良反应(AE)发生率没有明显的剂量相关性。
2.10 成熟抑制剂
HIV 成熟过程的重要一步是处理gag 蛋白。HIV蛋白酶将gag 蛋白切割成小块,产生结构性蛋白质,装配成具有传染性的成熟病毒粒子。MIs 通过结合gag 蛋白,干扰p24/p1 切割位点,使HIV 蛋白酶不能切割和处理gag 蛋白,导致无法形成成熟的病毒粒子。MIs 是一类很有潜力的新型分子,是具有全新作用机制的抗逆转录病毒 药物。
GSK3532795(BMS-955176)是一种在研的口服第2 代MIs,能在对现有药物耐药的HIV-1 感染者中起到关键作用。GSK3532795 对NRTIs、NNRTIs、PIs和INSTIs 耐药病毒具有活性,但A364V 突变与其高水平耐药相关。GSK3532795 单药治疗10 d,病毒载量下降可达到1.7 log10 拷贝/mL。AI468038 是一项随机双盲Ⅱb 期临床试验,206 例初治HIV-1 感染者按1 : 1 : 1 : 1 比例接受每日1 次GSK3532795(60、120、180 mg)或EFV 600 mg 每日1 次给药,并联合TDF/FTC 治疗。24 周分析数据显示,GSK3532795 和EFV与TDF/FTC 联合具有相似的疗效,GSK3532795 组和EFV 组分别有76% ~ 82.7% 和77.4% 的受试者获得病毒学抑制(HIV-1 RNA < 40 拷贝/mL)。GSK3532795 组和EFV组分别有2% ~ 8% 和17% 的受试者因不良反应停药,SAE 发生率分别为2% ~ 4% 和9%。然而,GSK3532795 组的胃肠道不良反应(主要是腹泻和腹痛)发生率为52% ~ 72.5%,明显高于EFV 组的发生率24.5%。GSK3532795组的NRTI 耐药发生率为6.5%,而EFV 组未发现NRTIs/NNRTIs 耐药。研究者将在今后的临床研究中进一步观察。
3 结语
30 年来,全球艾滋病的预防和治疗取得了巨大的成功。UNAIDS 提出在2020 年实现“90% 的艾滋病毒感染者被诊断、90% 的被诊断者接受治疗以及90% 的被治疗者获得病毒学抑制”的“90-90-90”目标,并力争在2030 年终结艾滋病流行的愿景。要实现这一伟大目标,仍然有漫长的路要走。目前已上市的抗HIV 药物还不能满足抗病毒治疗的需要,尤其是国内可供选择的抗病毒 药种类有限,多重耐药患者可能存在无药可用的困境,艾滋病的预防和治疗仍存在重大的临床需求。由于没有有效的**和根除HIV 病毒的方法,长期的抗逆转录病毒治疗仍然要面对药物的毒副作用、耐药病毒的产生和传播、患者依从性差、每日口服药物的不便利性等严峻挑战。药物研发的创新和进步对根除艾滋病至关重要,人类一直在积极探索新的治疗方法,寻找新的艾滋病药物靶点及针对该靶点的新药,特别是具有全新作用机制的药物。研发出高效、低毒、耐药屏障高、方便使用、依从性良好的抗HIV 药物是科学家们始终追求的目标。我们认为,以新复方单片制剂、长效注射药物、新作用机制药物、广谱中和抗体为代表的免疫治疗、无药缓解和功能性治愈将是未来抗艾滋病药物研发的热点和重点,也会在不久的将来取得重大突破。





合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2024 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2024 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57